A preprint version of the article is available at ChemRxiv.
Machine learning has transformed many fields and has recently found applications in chemistry and materials science. The small datasets commonly found in chemistry sparked the development of sophisticated machine learning approaches that incorporate chemical knowledge for each application and, therefore, require specialized expertise to develop. Here we show that GPT-3, a large language model trained on vast amounts of text extracted from the Internet, can easily be adapted to solve various tasks in chemistry and materials science by fine-tuning it to answer chemical questions in natural language with the correct answer. We compared this approach with dedicated machine learning models for many applications spanning the properties of molecules and materials to the yield of chemical reactions. Surprisingly, our fine-tuned version of GPT-3 can perform comparably to or even outperform conventional machine learning techniques, in particular in the low-data limit. In addition, we can perform inverse design by simply inverting the questions. The ease of use and high performance, especially for small datasets, can impact the fundamental approach to using machine learning in the chemical and material sciences. In addition to a literature search, querying a pre-trained large language model might become a routine way to bootstrap a project by leveraging the collective knowledge encoded in these foundation models, or to provide a baseline for predictive tasks.
One of the fascinating advances in machine learning has been the development of large language models (LLMs), so-called foundation models 1,2,3,4,5,6 . These models are appealing because of their simplicity; given a phrase, they return text that completes phrases in natural language such that, in many instances, one cannot tell that a machine wrote it.
From a scientific point of view, the most striking examples are that these foundation models can write sensible abstracts for scientific articles or even code for particular programming tasks 7,8,9,10,11,12 . Recently, it has been shown that these models can also solve relatively simple tabular regression and classification tasks 13 . However, as these models were not explicitly trained on these tasks, it is a remarkable result 5 .
That these models can solve simple tasks they are not trained for made us wonder whether they can also answer scientific questions for which we do not have an answer. As most chemistry problems can be represented in text form, we should be able to train these models to answer questions that chemists have. For example, ‘If I change the metal in my metal–organic framework, will it be stable in water?’ Such questions are often impossible to answer using theory or require highly sophisticated simulations or experiments.
We will always have very little (experimental) data for chemistry and material science applications. Hence, it is important that meaningful results can already be obtained with tens to hundreds of data points. We know from previous work on applications on text classification or generation that this works particularly well using models from the Generative Pre-trained Transformer 3 (GPT-3) family 5 , which were trained by the artificial intelligence company OpenAI. In this work, we show that these models—when provided with example data—perform surprisingly well for various chemistry questions, even outperforming the state-of-the-art machine learning models specifically developed for these tasks. It is important to realize that while language models have been used in chemistry before to predict properties 14,15,16,17 or design molecules 18,19,20 , they have conventionally been pre-trained on chemistry-specific tasks. In contrast, the models we investigate here have been trained on text corpi compiled mainly from the Internet but still can adapt to various tasks. Although ref. 8 has probed the inherent chemistry knowledge of LLMs, we focus on how those models perform when they are fine-tuned—that is, the weights are updated—on some task-specific dataset. Note that this task-specific fine-tuning makes the models less dependent on the prompt structure than in-context learning 21,22 .
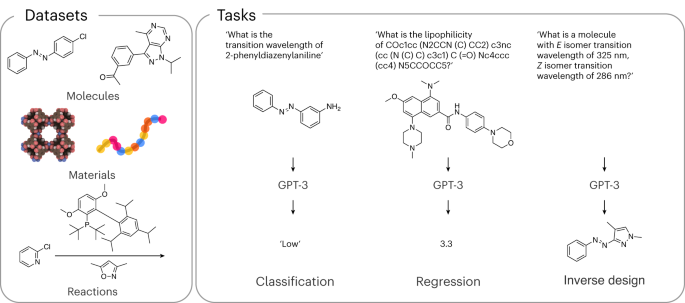
We benchmark our model on various datasets and applications to illustrate that these models can answer a wide range of scientific questions—ranging from the properties of materials, to how to synthesize materials and how to design materials (Fig. 1). In selecting these questions, we included some that have been addressed with machine learning. This allowed us to benchmark against state-of-the-art machine learning approaches specifically developed for these applications.
Before discussing the different applications in detail, let us first discuss how we fine-tune 23 the GPT-3 model in practice for a simple but highly non-trivial example. High-entropy alloys have attracted much interest as a novel class of structural metals. Interestingly, one has a sheer infinite number of possible combinations of metals. From a practical point of view, it is important to know whether a given combination of metals will form a solid solution or multiple phases. Hence, the question we would like to ask is: ‘What is the phase of ?’ and our model should give a text completion from the set of possible answers .
In Extended Data Table 1, we provide the set of questions and answers we used to fine-tune the GPT-3 model. These are questions and answers on high-entropy alloys for which the phase has been experimentally determined. The model tuning via the OpenAI API typically takes a few minutes and gives us a new model, which takes as input ‘Sm0.75Y0.25’ and gives as text completion ‘1’, which corresponds to single phase. This simple example already gives some remarkable results. We selected this example to directly compare its performance with the current state-the-art machine learning models with descriptors specially developed to mimic the relevant chemistry for this application 24 . In Fig. 2, we show that with only around 50 data points, we get a similar performance to the model of ref. 24 , which was trained on more than 1,000 data points.
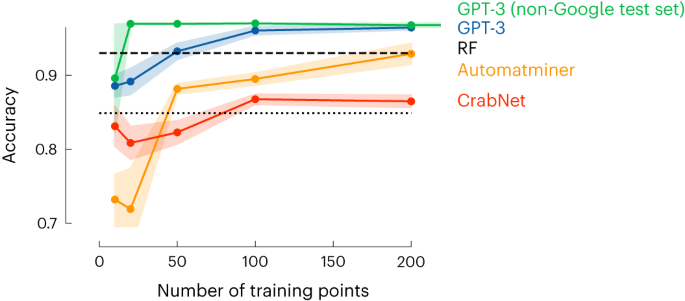
These results made us wonder whether similar results can be obtained for other properties. Hence, we looked at a range of very different properties of molecules, materials and chemical reactions. We focused on those applications for which conventional machine learning methods have been developed and generally accepted as benchmarks in their field. In addition, we also compared our model with the top-performing ones on tasks from the Matbench 25 suite of benchmarks (Supplementary Note 6.15).
Extended Data Table 2 compares the performance of a fine-tuned GPT-3 model with baselines (which can be found in Supplementary Note 6). For doing so, we fit the learning curves for the GPT-3 models and for the baselines and measure where they intersect, that is, we determine the factor of how much more (or fewer) data we would need to make the best baseline perform equal to the GPT-3 models in the low-data regime of the learning curves. The full learning curves for all models can be found in Supplementary Information (Supplementary Note 6).
For molecules, we investigated properties ranging from gaps between highest occupied (HOMO) and lowed unoccupied (LUMO) molecular orbitals and solubility in water to the performance in organic photovoltaics. For materials, we focused on the properties of alloys, metal–organic frameworks and polymers. Finally, for reactions, we considered two key cross-coupling reactions in organic chemistry. Extended Data Table 2 shows that in the low-data regime, our GPT-3 model is typically at least as good as the conventional machine learning model and often needs fewer data. In the high-data regime, the conventional machine learning models often catch up with the GPT-3 model. This makes sense, as for a given size of the dataset, the need for additional data and correlations (inductive biases) 26 captured by GPT-3 might be less needed.
We have to mention that we did not optimize the fine-tuning of the GPT-3 model, that is, we did not try to optimize how a sentence is presented to the model; one can envision that specific tokenization can have better results for chemical sentences 9,16,27,28 . Also, we did not tune the number of times we show an example to a model (that is, the number of epochs or the learning rate).
Importantly, we are also not limited to fine-tuning; in Supplementary Note 5, we show that we can even achieve good performance without fine-tuning by incorporating examples directly into the prompt (so-called in-context learning 5,29 , that is, learning during inference time). This works particularly well with the largest GPT-3 models and GPT-4. We are also not limited to using models from OpenAI. In Supplementary Notes 7 and 8, we also show that we could obtain good results by fine-tuning the open-source LLM’s parameter-efficient fine-tuning techniques on consumer hardware and provide a Python package that makes it easy to apply this approach to new problems.
An interesting question is how to represent a molecule or material. Most of the literature reports use International Union of Pure and Applied Chemistry (IUPAC) names. For machine learning applications, there has been a lot of effort to represent a chemical with unique line encodings (for example, simplified molecular-input line-entry system (SMILES) 30 or self-referencing embedded strings (SELFIES) 31,32 ). As the GPT-3 model has been trained on natural text, one might expect that chemical names are preferred over line representations such as SMILES or SELFIES. Therefore, we investigated different representations for our molecular property prediction tasks (see also Supplementary Note 4). Interestingly, our results (Supplementary Note 6) show that good results are obtained irrespective of the representation. The fact that we often get the best performance using the IUPAC name of the molecule makes fine-tuning GPT-3 for a particular application relatively simple for non-specialists.
A more challenging task than classification is to make a regression model, which would allow us to predict the value of a continuous property such as the Henry coefficient for the adsorption of a gas in a porous material. As we are using a pre-trained language model, performing actual regression that predicts real numbers ( \(\in <\mathbb
One can argue that the ultimate goal of machine learning in chemistry is to create a model that can generate molecules with a desired set of properties. This is also known as inverse design 33 . Broadly speaking, there are two approaches. If we have large datasets, we can train generative models such as variational autoencoders 34,35 or generative adversarial neural networks 36,37 . Without large datasets, evolutionary techniques such as genetic algorithms can generate novel, potentially interesting molecules 38,39,40,41 . Those evolutionary methods work best if one can limit the underlying chemistry; for example, finding the optimal functional group on a material with a well-defined backbone 42 .
Given that the GPT-3 model can predict the properties of molecules and materials with a small dataset, trying an inverse design strategy is tempting. This would be particularly important in the early stages of research; one often has a small set of experimental data points and a limited understanding. Yet, we could leverage a fine-tuned GPT-3 model to generate suggestions for novel materials with similar or even better performance. This would be an important step forward. Particularly as the tuning of such a natural language model is much more accessible than the training of conventional machine learning models. Here we investigate this setting: Can a fine-tuned GPT-3 propose valid molecules that satisfy the constraints or desired properties specified in a prompt in natural language? Again, we are illustrating the potential for a few case studies.
Molecular photoswitches are organic molecules with extended aromatic systems that make them responsive to light. Upon radiation, they switch reversibly between different isomers (which changes some properties, such as dipole moments). This reversible switching makes them interesting molecules for applications ranging from sensing to drug discovery. These molecules are complex, making sufficiently accurate predictions using first-principles theory very expensive. Yet, it is important to have some guidance to identify promising molecules, and machine learning models have been developed for this. One of the important properties of these photoswitches is the wavelength at which there is a maximum in the adsorption spectrum for the E and Z isomers. Hence, we fine-tuned GPT-3 with the same data used by ref. 43 . As we have shown above, we can fine-tune GPT-3 to accurately answer questions like ‘What is the pi–pi* transition wavelength of CN1C(/N=N/C2=CC=CC=C2)=C(C)C=C1C?’.
For GPT-3, inverse design is as simple as training the model with question and completion reversed. That is, answer the question ‘What is a photoswitch with transition wavelengths of 324 nm and 442 nm, respectively’ with a text completion that should be a SMILES string of a meaningful molecule. This approach should be contrasted with the approach used by ref. 43 , in which a library of molecules is generated, and their machine learning model (a Gaussian process regression) is used to evaluate the transition wavelengths of each material. If one has a lot of knowledge about the system, one can design large specific libraries that contain many promising molecules, including molecules with transition wavelengths of 324.0 nm and 442 nm. But, such a brute force technique is not what we understand as inverse design, as it, by definition, cannot predict a molecule that we did not include in our library.
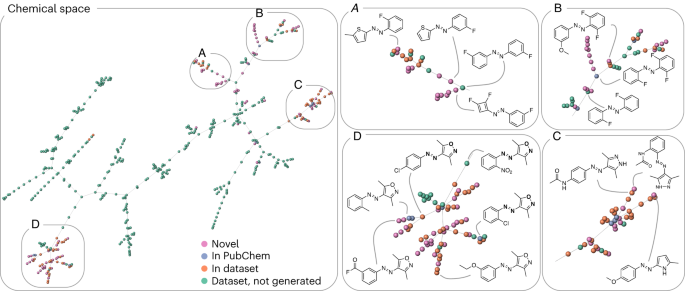
A simple test to see whether our model can generate new structures is to ask it to generate molecules with transition wavelengths similar to those from the dataset reported by ref. 43 . Extended Data Fig. 1 shows a representative sample of the molecules generated by the model. As expected, many molecules come from the training set (coloured orange in the figure). Importantly, many molecules are not in the training set, and, interestingly, some are not even in the PubChem database of known chemicals. In Fig. 3, we show that for the molecules, the transition wavelength is within a mean absolute percentage error of around 10%. Note that as the Gaussian process regression (GPR) model of ref. 43 was shown to perform comparably to, if not better than, more costly density functional theory simulations, we chose to use their model to compute the transition wavelengths for the generated molecules.
It is interesting to quantify how novel our newly generated molecules are. We compare these molecules to those collected in ref. 43 . We quantify the similarity by computing the distance between molecular fingerprints. Figure 4 visualizes this by laying out the resulting approximate nearest-neighbour graph in two dimensions. The orange and green spheres represent molecules from the ref. 43 dataset, the blue spheres show the novel ones, and the pink ones are not part of the PubChem database. As expected, we find many new structures that are derivatives of molecules in the ref. 43 database. However, we also find branches that are not part of the library of ref. 43 , indicating that the model generated novel kinds of compounds.
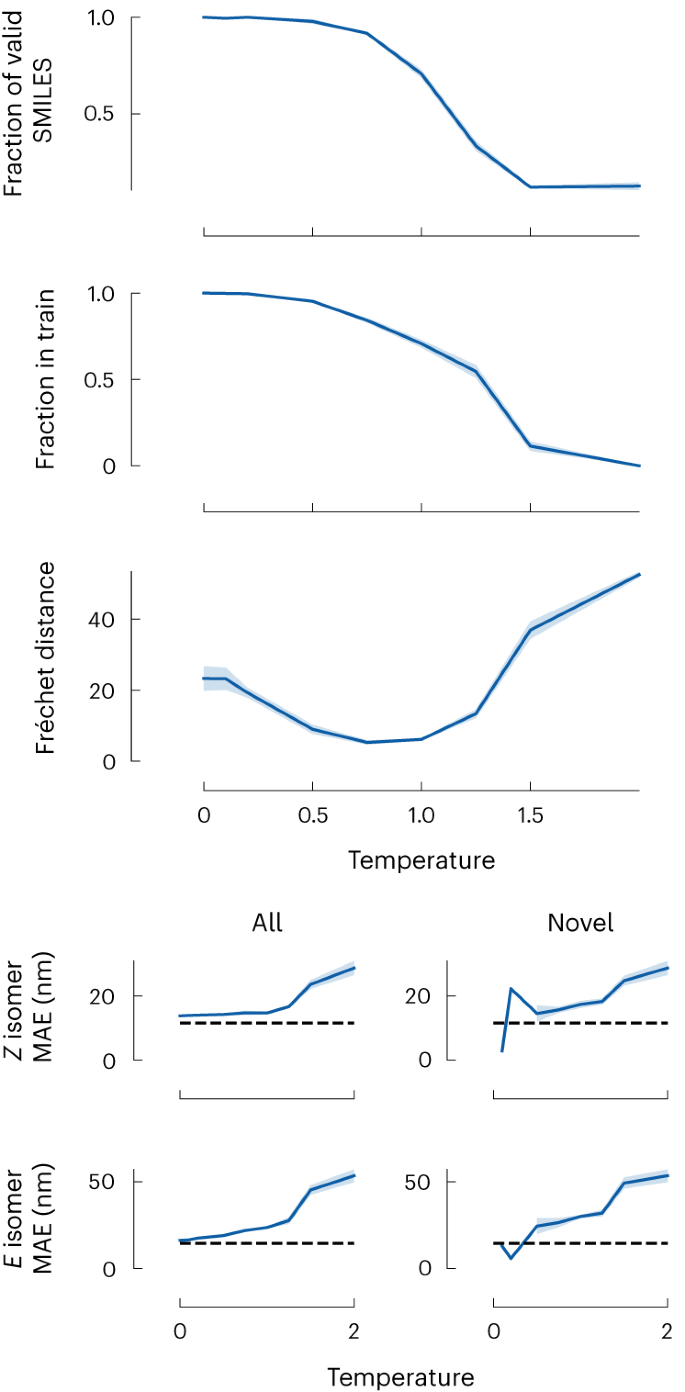
In generating these molecules, we adjusted the so-called softmax temperature in the sampling step of GPT-3 models. This temperature is conventionally used to generate more natural text. If we set this temperature to zero, we will generate text with the most frequently used words. We can increase the temperature to make the text more natural, making it more likely that less commonly used synonyms are chosen. For chemistry, if we aim to complete a SMILES starting with carbon, the zero-temperature solution would always complete the symbol that most commonly follows carbon (‘(’ in the QMugs dataset). In contrast, too-high temperatures would randomly choose any element.
The impact of this temperature parameter is shown in Fig. 3. At low temperatures, the generated molecules often come from the training set and only show a low diversity. Across all temperatures, the generated molecules seem synthesizable, as judged by a low synthetic accessibility (SA) score 44 . Increasing the temperature gives us more diverse and novel structures, but one can also expect more structures that make no chemical sense, that is, are invalid.
The results on the photoswitches illustrate the potential of LLMs for chemistry. To obtain more insight into whether we can trust these GPT-3 predictions, we carried out some experiments where we tried to stretch the limits.
We have already seen that we can obtain good results independent of how we represent a molecule (IUPAC names, SMILES or SELFIES), but can GPT-3 interpret an abstract representation of molecules we invented? A previous study 45 developed a machine learning approach to design dispersants using a coarse-grained approach. This dispersant was a linear copolymer with four monomer types and a chain length between 16 and 48 units, giving a chemical design space of 58 million different dispersants. One important goal in this work was to find dispersants with the right binding free energy, that is, which polymer length and which monomer sequence is optimal. As there is no way the GPT-3 model knows about the properties or representations of the coarse-grained polymers, it is interesting to see if we can get any sensible result if we ask the question ‘What is the adsorption free energy of coarse-grained dispersant AAAABBBBDDDDAAAACCCC’ or as inverse design, ‘Give me a structure of a coarse-grained dispersant with a free energy of 17’. Interestingly, for the prediction of the adsorption free energy, the GPT-3 model outperforms the models developed by ref. 45 . In addition, it can also successfully carry out the inverse design and generate monomer sequences that give the desired composition and, with a mean percentage error of around 22%, the desired adsorption free energy (the approximation of the ground truth we use already has a mean percentage error of around 9%, see Supplementary Note 11.1 for details).
In the case of the photoswitches, we have seen that the GPT-3 model can generate new molecules that are quite different from the training set. To explore in detail how far we can stretch the limits of what new molecules we can generate, we choose an application for which quantum calculations are known to predict the experimental values sufficiently accurately. The HOMO–LUMO gap is such an application. The HOMO–LUMO gap is relevant, for instance, in electronic applications that aim to excite a molecule at a specific energy. This HOMO–LUMO gap can be predicted accurately using semi-empirical quantum mechanics (GFN2-xTB 46 ), which is computationally affordable enough for us to compute for all generated molecules (Supplementary Note 77). Moreover, the QMugs dataset 47,48 has listed these HOMO–LUMO calculations for 665,000 molecules.
In Supplementary Note 11.3, we show that with the training of only 500 samples, we can get a reasonable estimate of the HOMO–LUMO gap of the molecules in the QMugs dataset. Also, by reverting the question, we have our model trained for inverse design. In Supplementary Note 11.3, we show that by asking the model ‘What is a molecule with a HOMO–LUMO gap of 3.5 eV’, we get similar to the photoswitches—a set of novel molecules. These novel molecules are not part of our training set and not even part of the QMugs dataset.
We now conduct some experiments on a dummy task to test how well the GPT-3 model can extrapolate to HOMO–LUMO gaps for which it has not received any training. To mimic this situation, we retrained our inverse design model using a dataset that has only molecules with HOMO–LUMO gaps smaller than 3.5 eV, and subsequently query the model with a question that requires the GPT-3 model to extrapolate (and, for example, to find that very small molecules are associated with large HOMO–LUMO gaps; a task we selected for only demonstration purposes and that can be exploited by generating small molecules). We do this by asking more than 1,000 times the question: ‘What is a molecule with a HOMO–LUMO gap of ’, where each time we slightly change the value of the HOMO–LUMO gap, that is, we sample XX from a Gaussian centred at 4 eV. Interestingly, the GPT-3 model does provide structures with a distribution of which our quantum calculations confirm that a meaningful fraction has a HOMO–LUMO gap >4.0 eV. Again, this is a remarkable result. In our training set, there was not a single molecule with a bandgap >3.5 eV, which shows that the GPT-3 model can make extrapolations. We can do a similar experiment for the photoswitches, for which we might have a library of photoswitches whose transition wavelengths are all below 350 nm. For practical applications, however, it can often be essential to have adsorption at larger wavelengths. In this case, we can successfully use a fine-tuned GPT-3 model to generate photoswitch molecules that adsorb at lower energy (Supplementary Fig. 75, which we also validated with time-dependent density functional theory in Supplementary Note 11.2.2).
These findings inspired us to do an inverse design experiment to design molecules with properties that take us far from the training set 49 . We are interested in molecules that have a HOMO–LUMO gap >5 eV. From the distribution of HOMO–LUMO gaps in the QMugs database (Fig. 5), we see that the average bandgap is around 2.58 eV. Only a handful of molecules in this database have a HOMO–LUMO gap above 5 eV.
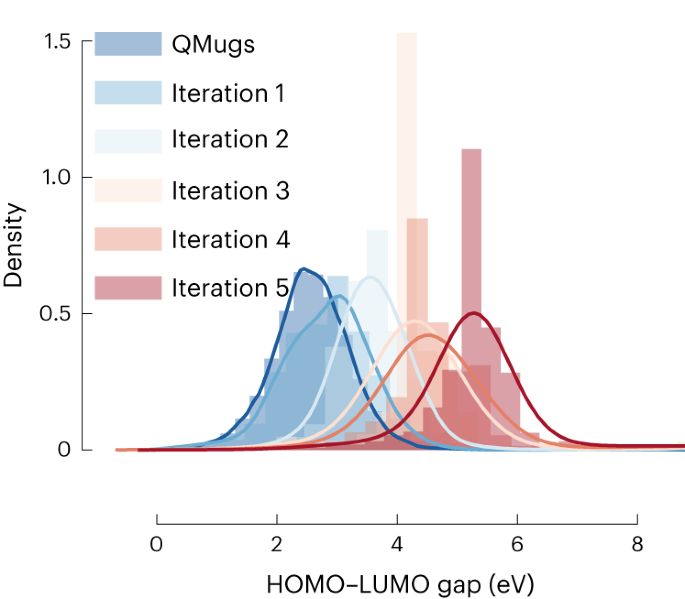
Hence, this is a challenging inverse design problem, as only a few materials in the database have the desired properties. Here our experiment is the quantum calculation, and we typically assume that we can evaluate hundreds to thousands of materials in a reasonable time. From a machine learning point of view, a set of thousands of materials is in a very low-data regime. However, from an experimental point of view, this is a large but sometimes doable effort. Of course, this is a somewhat arbitrary limit, and in Supplementary Fig. 83, we also give data for fewer experiments.
We start with the training using a set of hundreds of molecules randomly selected from the QMugs dataset (blue distribution in Fig. 5). These selected molecules will have bandgap distribution similar to the QMugs dataset. We then query for HOMO–LUMO gaps, now around 1,000 times requesting a molecule with a bandgap taken from a normal distribution with shifted mean (mean 4.0 eV, s.d. 0.2 eV). We evaluated these new molecules (green curve in Fig. 5), which indeed shows a shift of the distribution to higher HOMO–LUMO gaps. In the next iteration, we retrain the model with the new data and query again higher HOMO–LUMO gaps. Figure 5 shows that we have achieved our aim after four iterations.
Our results raise a very important question: how can a natural language model with no prior training in chemistry outperform dedicated machine learning models, as we were able to show in the case of high-entropy alloys in Fig. 2 and for various molecule, material and chemical reaction properties in Extended Data Table 2? To our knowledge, this fundamental question has no rigorous answer. The fact that we get good results independent of the chemical representation illustrates that these language models are very apt at extracting correlations from any text 15 . For example, we found promising results using both conventional chemical names and entirely hypothetical representations. In both cases, the model could quantitatively correlate the pattern of repeating units correctly to different kinds of properties.
Of course, if we say that the GPT-3 model is successful, it implies only that we have established that the GPT-3 model has identified correlations in the current training data that can be successfully exploited to make predictions. However, this does not imply that the correlations are always meaningful or related to cause–effect relationships. Hence, our research does not stop here. The next step will be to use GPT-3 to identify these correlations and ultimately get a deeper understanding. In this context, we argue that GPT-3 is only a tool to make more effective use of the knowledge scientists have collected over the years. It is also important to mention that while the training corpus contains chemistry information, many, if not most, scientific articles and results (including all failed or partially successful experiments 50 ) have not been seen by GPT-3. Hence, one can expect an even more impressive performance if these data are added to the training data.
As we show in this Article, a machine learning system built using GPT-3 works impressively well for a wide range of questions in chemistry—even for those for which we cannot use conventional line representations such as SMILES. Compared with conventional machine learning, it has many advantages. GPT-3 can be used for many different applications. Each application uses the same approach, in which the training and use of the model are based on questions formulated in natural language. This raises the bar for future machine learning studies, as any new models should at least outperform this simple approach instead.
The other important practical point is that using a GPT-3 model in a research setting is similar to a literature search. It will allow chemists to leverage the chemical knowledge we have collected. GPT-3 has been designed to discover correlations in text fragments, and the fact that these correlations are extremely relevant to chemistry opens many possibilities for chemists and material scientists alike.
For all the results shown in the main text, we used the smallest ada variant of GPT-3 available via the OpenAI API. For fine-tuning, we used the same setting for all case studies (8 epochs, learning rate multiplier of 0.02). Error bands show, if not otherwise indicated, the standard error of the mean.
To compare the data-efficiency of the GPT-3 models with our baselines, we fitted all learning curves to power laws (−a exp(−bx + c)). We then used these power laws to find where the best-performing baseline shows the same performance as the best GPT-3-based approach at the first learning curve point (that performs better than random, as measured using the Cohen’s kappa (κ) metric).
To check the validity of the generated SMILES we use the is_valid method from the Guacamol package 51 , which effectively considers a SMILES as valid if it can be parsed using RDKit.
We also performed some of our experiments by fine-tuning the GPT-J-6B model 52,53 (which has been trained on the Pile dataset 54 ) on consumer hardware using 8-bit quantization 55 and 8-bit optimizers 56 in addition to the low-rank adaptation (LoRA) technique 57 .
All data used in this work was obtained from public sources and can be downloaded from GitHub (https://github.com/kjappelbaum/gptchem) 58 .
All code created in this work is available on GitHub. The gptchem repository (https://github.com/kjappelbaum/gptchem) 58 contains all experiments with the OpenAI API. The chemlift repository (https://github.com/lamalab-org/chemlift) 59 contains an implementation supporting open-source LLMs.
K.M.J., A.O.-G. and B.S. were supported by the MARVEL National Centre for Competence in Research funded by the Swiss National Science Foundation (grant agreement ID 51NF40-182892). P.S. acknowledges support from NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. The research of K.M.J. and B.S. was also supported by the USorb-DAC Project, which is funded by a grant from The Grantham Foundation for the Protection of the Environment to RMI’s climate tech accelerator programme, Third Derivative. In addition, the work of K.M.J. was supported by the Carl-Zeiss Foundation.
Open access funding provided by EPFL Lausanne.